Fragment Processing#
Setup#
Download the PBMC 5k ATAC-seq dataset from 10x Genomics if you haven’t already:
%%bash
[ -f data/pbmc_fragments.tsv.gz ] || wget -O data/pbmc_fragments.tsv.gz https://cf.10xgenomics.com/samples/cell-atac/1.2.0/atac_pbmc_5k_nextgem/atac_pbmc_5k_nextgem_fragments.tsv.gz
[ -f data/gencode.v44.primary_assembly.annotation.gtf.gz ] || wget -O data/gencode.v44.primary_assembly.annotation.gtf.gz http://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_44/gencode.v44.primary_assembly.annotation.gtf.gz
import gatac as ga
import matplotlib.pyplot as plt
import time
import numpy as np
print(f"GATAC version: {ga.__version__}")
GATAC version: 0.1.0
1. Convert TSV.GZ to Parquet#
GATAC uses Parquet as its internal fragment format. Parquet stores data in row groups that can be read independently, enabling GPU-streaming over files larger than available VRAM.
FRAGMENTS_TSV = "data/pbmc_fragments.tsv.gz"
t0 = time.perf_counter()
parquet_path = ga.pp.make_parquet(FRAGMENTS_TSV,barcode_prefix="sample1")
print(f"Converted in {time.perf_counter() - t0:.1f}s → {parquet_path}")
15:18:57 - gatac.pp.convert - INFO - Converting pbmc_fragments.tsv.gz to Parquet
15:19:13 - gatac.pp.convert - INFO - Created pbmc_fragments.parquet
Converted in 15.9s → data/pbmc_fragments.parquet
The equivalent cli command is:
gatac convert data/pbmc_fragments.tsv.gz --barcode-prefix sample1
By default, this saves the Parquet file to data/pbmc_fragments.parquet. The --barcode-prefix option is useful when processing multiple independent samples to prevent barcode collisions.
2. Fragment size distribution#
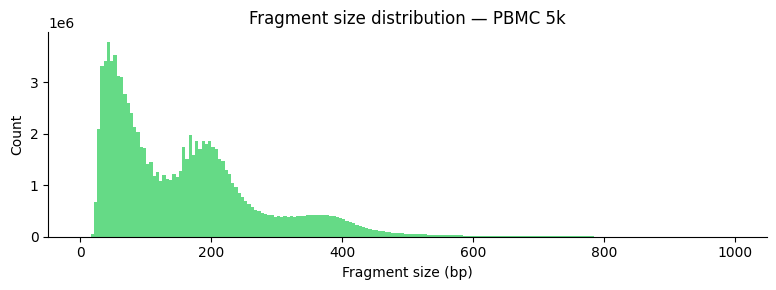
ATAC-seq fragment sizes show a characteristic nucleosomal ladder pattern: sub-nucleosomal (<150 bp), mono-nucleosomal (~200 bp), di-nucleosomal (~400 bp).
ga.pl.fragment_size_distribution(parquet_path, title="Fragment size distribution — PBMC 5k")
<Axes: title={'center': 'Fragment size distribution — PBMC 5k'}, xlabel='Fragment size (bp)', ylabel='Count'>
3. Quality metrics#
GTF = "data/gencode.v44.primary_assembly.annotation.gtf.gz"
t0 = time.perf_counter()
metrics = ga.pp.compute_metrics(
parquet_path,
GTF,
window_size=2000,
smooth_window=11,
min_unique_frags=500,
exclude_chroms=["chrM", "M"],
row_groups_per_batch=64,
)
print(f"Computed metrics for {len(metrics):,} barcodes in {time.perf_counter() - t0:.1f}s")
metrics_pd = metrics.to_pandas()
15:19:16 - gatac.pp.metrics - INFO - Loading TSS from data/gencode.v44.primary_assembly.annotation.gtf.gz (Polars)
15:19:18 - gatac.pp.metrics - INFO - Loaded 219,362 unique TSSs (Polars)
15:19:18 - gatac.pp.metrics - INFO - Computing metrics (streaming mode, row_groups_per_batch=64)
15:19:18 - gatac.pp.metrics - INFO - Aggregating QC metrics from 101 row groups in batches of 64...
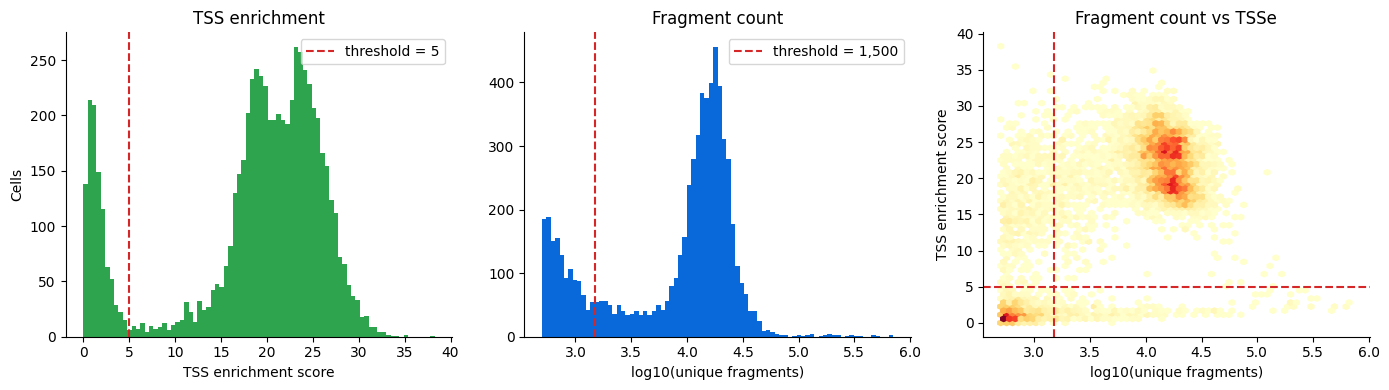
15:19:20 - gatac.pp.metrics - INFO - Found 6,604 cells with >= 500 unique fragments
15:19:20 - gatac.pp.metrics - INFO - Processing 101 row groups in batches of 64...
15:19:22 - gatac.pp.metrics - INFO - Processed row groups 101/101
15:19:22 - gatac.pp.metrics - INFO - Completed TSSe computation
Computed metrics for 6,604 barcodes in 3.8s
The equivalent cli command would be:
gatac metrics data/pbmc_fragments.parquet \
-g data/gencode.v44.primary_assembly.annotation.gtf.gz
Saving metrics to data/pbmc_fragments_metrics.csv by default.
ga.pl.qc_metrics(metrics, tsse_threshold=5, n_unique_threshold=1500)
4. Filter QC & Build tile matrix#
We chose to retain the fragment file containing all cell barcodes, subsequently filtering out low-quality cells while binning the genome into 500 bp tiles. Unique fragment insertions per cell were then quantified using the SnapATAC2-compatible "unique" counting strategy.
t0 = time.perf_counter()
adata = ga.pp.make_tile_matrix(
parquet_path,
chrom_sizes="hg38",
metrics=metrics,
filter_query="(n_unique >= 1500) & (tsse_score >= 5)",
tile_size=500,
exclude_chroms=["chrM", "chrY"],
count_strategy="unique",
)
print(f"Tile matrix built in {time.perf_counter() - t0:.1f}s")
print(adata)
15:19:36 - gatac.pp.tile - INFO - Processing pbmc_fragments.parquet
15:19:56 - gatac.pp.tile - INFO - Created pbmc_fragments_tile_matrix.h5ad: 5,001 cells × 6,062,095 tiles (20.3s)
Tile matrix built in 20.4s
AnnData object with n_obs × n_vars = 5001 × 6062095
obs: 'barcode', 'n_unique', 'tsse_score', 'duplicate_fraction', 'mito_fraction'
var: 'chrom', 'start', 'end'
The equivalent CLI command is:
gatac tile data/pbmc_fragments.parquet -g hg38 \
--metrics data/pbmc_fragments_metrics.csv \
--filter "(n_unique >= 1500) & (tsse_score >= 5)"
By default, the tile matrix will be saved to data/pbmc_fragments_tile_matrix.h5ad.
If you prefer to filter the parquet file before running the tiling process, use the following commands:
gatac filter data/pbmc_fragments.parquet \
--metrics data/pbmc_fragments_metrics.csv \
--filter "(n_unique >= 1500) & (tsse_score >= 5)"
gatac tile data/pbmc_fragments_filtered.parquet
# Summary stats
print(f"Cells: {adata.n_obs:,}")
print(f"Tiles: {adata.n_vars:,}")
print(f"Density: {adata.X.nnz / (adata.n_obs * adata.n_vars) * 100:.2f}%")
Cells: 5,001
Tiles: 6,062,095
Density: 0.29%
5. Feature selection#
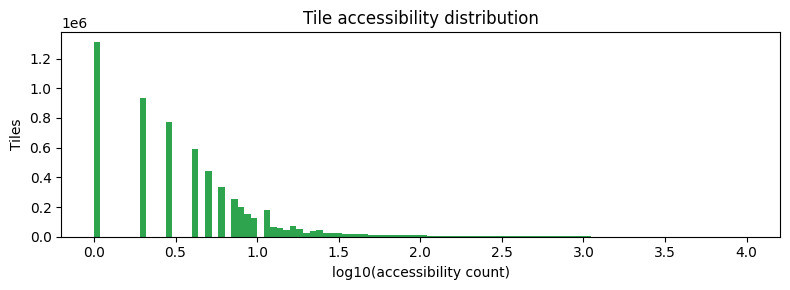
feature_counts = np.asarray(adata.X.sum(axis=0)).flatten()
fig, ax = plt.subplots(figsize=(8, 3))
ax.hist(np.log10(feature_counts + 1), bins=100, color="#2ea44f", edgecolor="none")
ax.set_xlabel("log10(accessibility count)")
ax.set_ylabel("Tiles")
ax.set_title("Tile accessibility distribution")
plt.tight_layout()
plt.show()
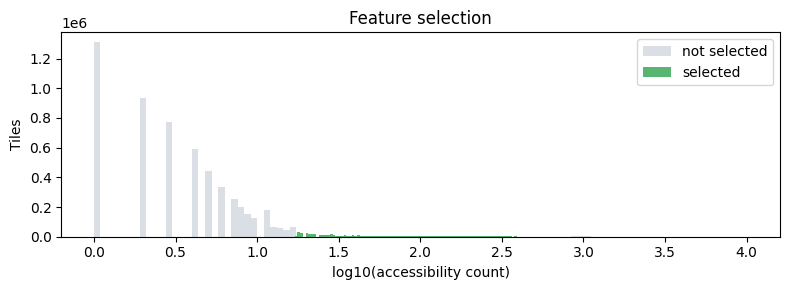
select_features keeps the top n_features most accessible tiles after
removing very rare (<0.5th percentile) and ubiquitously open (>99.5th
percentile) features — following the ArchR strategy.
t0 = time.perf_counter()
ga.pp.select_features(
adata,
n_features=500_000,
)
print(f"Feature selection in {time.perf_counter() - t0:.1f}s")
n_selected = adata.var["selected"].sum()
print(f"Selected {n_selected:,} / {adata.n_vars:,} features")
15:19:59 - gatac.pp.features - INFO - Selecting features from 6,062,095 total
15:19:59 - gatac.pp.features - INFO - Detected count matrix - using quantile-filtered selection
15:20:00 - gatac.pp.features - INFO - Selected 500,000 features
Feature selection in 0.7s
Selected 500,000 / 6,062,095 features
The equivalent cli command would be:
gatac features data/pbmc_fragments_tile_matrix.h5ad
Saving to data/pbmc_fragments_tile_matrix_selected.h5ad by default.
Note that this function accept a list of tile matrices (if processing multiple samples). The output filtered tile matrices can be combined with:
gatac features sample*.h5ad
gatac combine sample*tile_matrix.h5ad -o combined.h5ad
In that case, make sure to have used barcode-prefix in the previous steps to prevent barcode collision.
# Highlight selected features
fig, ax = plt.subplots(figsize=(8, 3))
is_selected = adata.var["selected"].values
counts = adata.var["accessibility_count"].values
ax.hist(np.log10(counts[~is_selected] + 1), bins=100,
color="#d0d7de", edgecolor="none", label="not selected", alpha=0.8)
ax.hist(np.log10(counts[is_selected] + 1), bins=100,
color="#2ea44f", edgecolor="none", label="selected", alpha=0.8)
ax.set_xlabel("log10(accessibility count)")
ax.set_ylabel("Tiles")
ax.set_title("Feature selection")
ax.legend()
plt.tight_layout()
plt.show()
6. Save#
adata.write_h5ad("data/pbmc_tile.h5ad")
print("Saved → data/pbmc_tile.h5ad")
Saved → data/pbmc_tile.h5ad
Next steps#
02 – Spectral embedding: dimensionality reduction and clustering